Kepivance: Package Insert / Prescribing Info
Package insert / product label
Generic name: palifermin
Dosage form: injection, powder, lyophilized, for solution
Drug class: Miscellaneous uncategorized agents
J Code (medical billing code): J2425 (50 mcg, injection)
Medically reviewed by Drugs.com. Last updated on Dec 17, 2024.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
KEPIVANCE ®(palifermin) for injection, for intravenous use
Initial U.S. Approval: 2004
Indications and Usage for Kepivance
- Kepivance is a mucocutaneous epithelial human growth factor indicated to decrease the incidence and duration of severe oral mucositis in patients with hematologic malignancies receiving myelotoxic therapy in the setting of autologous hematopoietic stem cell support. Kepivance is indicated as supportive care for preparative regimens predicted to result in ≥ WHO Grade 3 mucositis in the majority of patients. ( 1.1)
Limitations of Use
- The safety and efficacy of Kepivance have not been established in patients with non-hematologic malignancies ( 1.2, 5.1)
- Kepivance was not effective in decreasing the incidence of severe mucositis in patients with hematologic malignancies receiving myelotoxic therapy in the setting of allogeneic hematopoietic stem cell support. ( 1.2, 14.3)
- Kepivance is not recommended for use with melphalan 200 mg/m 2as a conditioning regimen ( 14.2).
Kepivance Dosage and Administration
Administer as an intravenous bolus injection at a dose of 60 mcg/kg/day for 3 consecutive days before and 3 consecutive days after myelotoxic therapy for a total of 6 doses ( 2.1)
- Administer the first 3 doses prior to myelotoxic therapy with the third dose 24 to 48 hours before myelotoxic therapy ( 2.1)
- Administer the last 3 doses after myelotoxic therapy is complete with the first of these doses on the day of hematopoietic stem cell infusion after the infusion is completed, and at least 7 days after the most recent administration of Kepivance ( 2.1)
Dosage Forms and Strengths
For injection: 5.16 mg lyophilized powder in single-dose vials ( 3)
Contraindications
None (4)
Warnings and Precautions
Adverse Reactions/Side Effects
Most common adverse reactions (incidence ≥20% and 5%≥placebo) are rash, fever, elevated serum amylase, pruritus, erythema, and edema ( 6)
To report SUSPECTED ADVERSE REACTIONS, contact Swedish Orphan Biovitrum at 1-866-773-5274 or FDA at 1-800-FDA-1088 orwww.fda.gov/medwatch.
Drug Interactions
Use In Specific Populations
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 7/2023
Full Prescribing Information
1. Indications and Usage for Kepivance
1.1 Indications
Kepivance is indicated to decrease the incidence and duration of severe oral mucositis in patients with hematologic malignancies receiving myelotoxic therapy in the setting of autologous hematopoietic stem cell support. Kepivance is indicated as supportive care for preparative regimens predicted to result in ≥ WHO Grade 3 mucositis in the majority of patients. Kepivance is a mucocutaneous epithelial human growth factor indicated to decrease the incidence and duration of severe oral mucositis in patients with hematologic malignancies receiving myelotoxic therapy in the setting of autologous hematopoietic stem cell support. Kepivance is indicated as supportive care for preparative regimens predicted to result in ≥ WHO Grade 3 mucositis in the majority of patients.
1.2 Limitations of Use
The safety and efficacy of Kepivance have not been established in patients with non-hematologic malignancies )]. The safety and efficacy of Kepivance have not been established in patients with non-hematologic malignancies [see Warnings and Precautions ( 5.1)].
Kepivance was not effective in decreasing the incidence of severe mucositis in patients with hematologic malignancies receiving myelotoxic therapy in the setting of allogeneic hematopoietic stem cell support. Kepivance was not effective in decreasing the incidence of severe mucositis in patients with hematologic malignancies receiving myelotoxic therapy in the setting of allogeneic hematopoietic stem cell support [See Clinical Studies ( 14.3)] .
Kepivance is not recommended for use with melphalan 200 mg/m as a conditioning regimen. Kepivance is not recommended for use with melphalan 200 mg/m 2as a conditioning regimen [See Clinical Studies ( 14.2)] .
2. Kepivance Dosage and Administration
2.1 Recommended Dosage Regimen
The recommended dose of Kepivance is 60 mcg/kg/day, administered as an intravenous bolus injection for 3 consecutive days before and 3 consecutive days after myelotoxic therapy, for a total of 6 doses. The recommended dose of Kepivance is 60 mcg/kg/day, administered as an intravenous bolus injection for 3 consecutive days before and 3 consecutive days after myelotoxic therapy, for a total of 6 doses.
Administer the first 3 doses prior to myelotoxic therapy. Administer the third dose 24 to 48 hours prior to beginning myelotoxic therapy Administer the first 3 doses prior to myelotoxic therapy. Administer the third dose 24 to 48 hours prior to beginning myelotoxic therapy [see Drug Interactions ( 7)].
Administer the last 3 doses after myelotoxic therapy is complete; administer the first of these doses on the day of hematopoietic stem cell infusion after the infusion is completed, and at least 7 days after the most recent administration of Kepivance [see Drug Interactions ( 7)].
2.2 Preparation and Administration
Preparation
Prepare the solution for infusion, using aseptic technique, as follows:
- Reconstitute Kepivance lyophilized powder with Sterile Water for Injection, USP (not supplied) by slowly injecting 1.2 mL of Sterile Water for Injection, USP to yield a final concentration of 5 mg/mL.
- Swirl the contents gently during dissolution. Do not shake or vigorously agitate the vial. Dissolution of Kepivance can take up to 3 minutes.
- Visually inspect the solution for discoloration and particulate matter before administration. The reconstituted solution should be clear and colorless. Do not administer Kepivance if discoloration or particulates are observed. Do not filter the reconstituted solution during preparation or administration. Do not freeze the reconstituted solution. Protect from light.
Administration
- Administer Kepivance by intravenous bolus injection. If heparin is used to maintain an intravenous line, rinse the line with saline prior to and after Kepivance administration [see Drug Interactions ( 7)].
- The reconstituted solution contains no preservatives and is intended for single use only. Discard any unused portion.
- Following reconstitution, it is recommended that the product be used immediately. If not used immediately, the reconstituted solution of Kepivance may be stored refrigerated in its carton at 2° to 8°C (36° to 46°F) for up to 24 hours.
- Prior to injection, allow Kepivance to reach room temperature for a maximum of 1 hour protected from light. Discard Kepivance left at room temperature for more than 1 hour.
5. Warnings and Precautions
5.1 Potential for Stimulation of Tumor Growth
The safety and efficacy of Kepivance have not been established in patients with non-hematologic malignancies. The effects of Kepivance on stimulation of KGF receptor-expressing, non-hematopoietic tumors in patients are not known. Kepivance has been shown to enhance the growth of human epithelial tumor cell lines in vitroand to increase the rate of tumor cell line growth in a human carcinoma xenograft model [see Clinical Pharmacology ( 12.1)].
6. Adverse Reactions/Side Effects
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The data described in Table 1and the discussion below reflect exposure to Kepivance in 409 patients with hematologic malignancies who were enrolled in 3 randomized, placebo-controlled clinical trials and a pharmacokinetic study. Patients received Kepivance either before, or before and after, regimens of myelotoxic chemotherapy, with or without total body irradiation (TBI), followed by hematopoietic stem cell support. Kepivance was administered in daily doses ranging from 5 to 80 mcg/kg/day. The total dose of Kepivance ranged from 15 to 480 mcg/kg with a median of 360 mcg/kg. The population had a median age of 48 years (range: 41 to 60 years), 62% were male and 83% were White with 7.4 % Black and 6.2 % Hispanic. Non Hodgkin's lymphoma (NHL) was the most common malignancy followed by Hodgkin's disease, multiple myeloma, and leukemia.
The most common adverse reactions attributed to Kepivance were skin toxicities (rash, erythema, edema, pruritus), oral toxicities (dysesthesia, tongue discoloration, tongue thickening, alteration of taste), pain, arthralgias, and dysesthesia. The median time to onset of cutaneous toxicity was 6 days following the first of 3 consecutive daily doses of Kepivance, with a median duration of 5 days. In patients receiving Kepivance, dysesthesia (including hyperesthesia, hypoesthesia, and paresthesia) was usually localized to the perioral region, whereas in patients receiving placebo dysesthesias were more likely to occur in extremities.
The most common serious adverse reaction attributed to Kepivance was skin rash, reported in less than 1% (3/409) of patients treated. Grade 3 skin rashes occurred in 3% of patients (9/409) receiving Kepivance and 2% (5/241) receiving placebo.
| BODY SYSTEM
Adverse Reaction | Kepivance
(n = 409) % | Placebo
(n = 241) % |
| BODY AS A WHOLE | ||
| Edema | 28 | 21 |
| Pain | 16 | 11 |
| Fever | 39 | 34 |
| GASTROINTESTINAL | ||
| Mouth/Tongue Thickness or Discoloration | 17 | 8 |
| MUSCULOSKELETAL | ||
| Arthralgia | 10 | 5 |
| SKIN AND APPENDAGES | ||
| Rash | 62 | 50 |
| Pruritus | 35 | 24 |
| Erythema | 32 | 22 |
| SPECIAL SENSES | ||
| Taste Altered | 16 | 8 |
| CENTRAL NERVOUS SYSTEM / PERIPHERAL NERVOUS SYSTEM | ||
| Dysesthesia – Hyperesthesia / hypoesthesia/ paresthesia | 12 | 7 |
| METABOLIC | ||
| Elevated serum lipase
| ||
| All grades | 28 | 23 |
| Grade 3 and 4 | 11 | 5 |
| Elevated serum amylase | ||
| All grades | 62 | 54 |
| Grade 3 and 4 | 38 | 31 |
Cataracts:In a postmarketing safety study, the incidence of cataracts was numerically higher among patients receiving Kepivance than in the control population. (See 14 CLINICAL STUDIES).
Infections:In a randomized, double-blind, placebo-controlled post-approval study designed to determine the efficacy of Kepivance with a high-dose melphalan preparative regimen, the incidence of treatment-emergent infections was significantly greater in patients treated with Kepivance compared to placebo. A total of 281 patients were randomized to 3 arms: Kepivance before melphalan on days -6, -5, -4 and after melphalan on days 0, 1, and 2 (pre-post) (n=115); Kepivance before melphalan on days -6, -5, -4 (pre) (n=109); or placebo (n=57). The incidence of reported infections were pre-post - 50%; pre - 47%; and placebo - 25% [see Clinical Studies ( 14.2)] .
Laboratory Test Findings:Reversible elevations in serum lipase and amylase, which did not require treatment, were reported in 28% and 62% of patients receiving Kepivance and 23% and 54% of patients receiving placebo. In general, peak increases were observed during the period of cytotoxic therapy and returned to baseline by the day of hematopoietic stem cell infusion. Amylase was mainly salivary in origin.
6.2 Immunogenicity
As with all therapeutic proteins, there is a potential for immunogenicity. The clinical significance of antibodies to Kepivance is unknown but may include decreased activity and/or cross reactivity with other members of the FGF family of growth factors.
In clinical trials, serum samples from patients treated with Kepivance were tested for antibodies to Kepivance using an electrochemiluminescence-based binding assay. Twelve of 645 patients (2%) tested positive; none had evidence of neutralizing activity in a cell-based assay.
The incidence of antibody positivity is highly dependent on the specific assay and its sensitivity. Additionally, the observed incidence of antibody positivity in an assay may be influenced by several factors including sample handling, timing of sample collection, concomitant medications and underlying disease. For these reasons, comparison of the incidence of antibodies to Kepivance with the incidence of antibodies to other products may be misleading.
6.3 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of Kepivance in the stem cell transplant setting. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Vaginal edema and erythema;
- Palmar-plantar Erythrodysaesthesia Syndrome (also known as “hand-foot syndrome”)
7. Drug Interactions
In vitroand in vivodata showed that palifermin interacts with unfractionated as well as low molecular weight heparins with no noticeable effect on the pharmacodynamics of either drug. If heparin is used to maintain an intravenous line, rinse the line with saline prior to and after Kepivance administration [see Clinical Pharmacology ( 12.3)] .
Do not administer Kepivance within 24 hours before, during infusion of, or within 24 hours after administration of myelotoxic chemotherapy [see Dosage and Administration ( 2.1) and Clinical Studies ( 14)]. In a clinical trial, administration of Kepivance within 24 hours of chemotherapy resulted in increased severity and duration of oral mucositis.
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Based on findings in animal studies, Kepivance may cause fetal harm when administered to pregnant women. There are no data available on Kepivance use in pregnant women to inform a drug-associated risk of major birth defects and miscarriage or adverse maternal or fetal outcomes. In animal reproduction studies, intravenous administration of palifermin to pregnant rabbits and rats during the period of organogenesis resulted in embryo-fetal mortality and alterations to growth [see Data] .
The estimated background risk of major birth defects and miscarriage for the indicated populations is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Data
Animal data
In embryo-fetal development studies, palifermin was administered intravenously to pregnant rabbits and rats during the perid of organogenesis. Doses were 5, 60, and 150 μg/kg/day in rabbits (gestation days 6-18) and 100, 300, and 1000 μg/kg/day in rats (gestation days 6 through 17). Increased post-implantation loss and decreased fetal body weights occurred along with maternal toxicity (clinical signs and reductions in body weight gain and food consumption) at doses of 150 μg/kg/day in rabbits and 1000 μg/kg/day in rats. Increased skeletal variations was noted in rats at 1000 μg/kg/day. Doses of 150 μg/kg/day in rabbits and 1000 μg/kg/day in rats are approximately 5-times (rabbits) and 35-times (rats) the exposure (AUC) in patients receiving the recommended dose of 60 μg/kg/day.
8.2 Lactation
Risk Summary
There are no data on the presence of Kepivance in human milk, the effect on the breastfed child, or the effect on milk production. Since many drugs are secreted into human milk, and because of the potential for serious adverse reactions in a nursing child, breastfeeding should be discontinued during treatment and for at least 2 weeks after the last dose.
8.3 Females and Males of Reproductive Potential
Infertility
Based on findings from animal studies, palifermin may impair fertility in females and males of reproductive potential [see Nonclinical Toxicology ( 13.1)] . The reversibility of the effects on fertility is unknown.
8.4 Pediatric Use
Information on the dosing and safety of Kepivance in the pediatric population is limited. However, use of Kepivance in pediatric patients ages 1 to 16 years is supported by evidence from adequate and well-controlled studies of Kepivance in adults and a phase 1 study that included 27 pediatric patients with acute leukemia undergoing hematopoietic stem cell transplant. Three age groups were studied: ages 1 to 2 (n=9), ages 3 to 11 (n=9), and ages 12 to 16 (n=9); 56% were male, 26% were Caucasian, 63% Hispanic; 81% ALL, 19% AML. The patients received high-dose cytotoxic therapy consisting of fractionated total body irradiation (TBI) (12 Gy total dose), high dose etoposide (1500 mg/m 2), and high dose cyclophosphamide (120 mg/kg) followed by allogeneic hematopoietic stem cell support. The dose intensity of this preparative regimen is comparable to the dose intensity of the Study 1 preparative regimen. See Clinical Studies [14.1]. Kepivance was administered as a daily intravenous injection for 3 consecutive days prior to initiation of cytotoxic therapy and for 3 consecutive days following infusion of hematopoietic stem cells. Three dose levels, 40, 60, and 80 mcg/kg/dose, were evaluated. There was no dose limiting toxicity identified at any dose level. Adverse events were similar to those reported in adult studies. The incidence of palifermin related adverse events was highest in the 80 μg/kg cohort. The overall incidence of WHO grade 3 and 4 oral mucositis was 10/27 (37%).
The pharmacokinetics of Kepivance was evaluated in the phase 1 study. Age (1 to 16 years) did not affect the pharmacokinetics of palifermin over the dose range (40 to 80 mcg/kg). Palifermin concentrations declined in the first 30 minutes after dosing. An increase in palifermin concentrations occurred at around 2 to 4 hours post-dose for some subjects, which was followed by a second, slow decline phase. A similar trend has been observed in adult patients. The mean half-life range was 2.6 to 5.6 hours in pediatric patients following the first 60 mcg/kg dose of Kepivance. No accumulation was observed following 3 consecutive doses of Kepivance. Palifermin exposure did not increase linearly with increasing doses. The first dose AUC 0-inf(mean) of Kepivance 60 mcg/kg/day in adult patients (18 to 63 years) was 38.2 ng*hr/mL compared to 46.1 ng*hr/mL (range of means: 22.8 to 81.6) for pediatric patients (1 to 16 years). The mean clearance was 1730 mL/hr/kg for adults and 2481 mL/hr/kg (range of means: 1700 to 3460) in pediatric patients.
8.5 Geriatric Use
Clinical studies of Kepivance did not include sufficient numbers of subjects aged 65 years and older to determine whether they responded differently from younger subjects [see Clinical Pharmacology ( 12.3)].
11. Kepivance Description
Kepivance (palifermin) is a truncated human KGF produced by recominant DNA technology in E coli. Kepivance is a water soluble, 140 amino acid protein with a molecular weight of 16.3 kilodaltons. It differs from endogenous human KGF in that the first 23 N terminal amino acids have been deleted to improve protein stability.
Kepivance is supplied as a sterile, white, preservative-free, lyophilized powder for intravenous injection after reconstitution with 1.2 mL of Sterile Water for Injection, USP. Reconstitution yields a clear, colorless solution of Kepivance (5 mg/mL) with a pH of 6.5. Each single-dose vial of Kepivance contains palifermin (5.16 mg),with L histidine (1.60 mg), mannitol (41 mg), polysorbate 20 (0.11 mg or 0.01% w/v), and sucrose (21 mg).
12. Kepivance - Clinical Pharmacology
12.1 Mechanism of Action
KGF is an endogenous protein in the fibroblast growth factor (FGF) family that binds to the KGF receptor. Binding of KGF to its receptor has been reported to result in proliferation, differentiation, and migration of epithelial cells. The KGF receptor, one of four receptors in the FGF family, has been reported to be present on epithelial cells in many tissues examined including the tongue, buccal mucosa, esophagus, stomach, intestine, salivary gland, lung, liver, pancreas, kidney, bladder, mammary gland, skin (hair follicles and sebaceous gland), and the lens of the eye. The KGF receptor has been reported to not be present on cells of the hematopoietic lineage. Endogenous KGF is produced by mesenchymal cells and is upregulated in response to epithelial tissue injury.
In mice and rats, Kepivance enhanced proliferation of epithelial cells (as measured by Ki67 immunohistochemical staining and BrDU uptake) and demonstrated an increase in tissue thickness of the tongue, buccal mucosa, and gastrointestinal tract. Kepivance has been studied in murine models of chemotherapy and radiation-induced gastrointestinal injury. In such models, administration of Kepivance prior to and/or after the cytotoxic insult improved survival and reduced weight loss compared to control animals.
Kepivance has been shown to enhance the growth of human epithelial tumor cell lines in vitroat concentrations ≥ 10 mcg/mL (> 15-fold higher than average therapeutic concentrations in humans). In nude mouse xenograft models, three consecutive daily treatments of Kepivance at doses of 1,500 and 4,000 mcg/kg (25- and 67-fold higher than the recommended human dose, respectively) repeated weekly for 4 to 6 weeks were associated with a dose-dependent increase in the growth rate of 1 of 7 KGF receptor-expressing human tumor cell lines.
12.2 Pharmacodynamics
Epithelial cell proliferation was assessed by Ki67 immunohistochemical staining in healthy subjects. A 3-fold or greater increase in Ki67 staining was observed in buccal biopsies from 3 of 6 healthy subjects given Kepivance at 40 mcg/kg/day intravenously for 3 days, when measured 24 hours after the third dose. Dose-dependent epithelial cell proliferation was observed in healthy subjects given single intravenous doses of 120 to 250 mcg/kg 48 hours post-dosing.
12.3 Pharmacokinetics
The pharmacokinetics of Kepivance were studied in healthy subjects and patients with hematologic malignancies. After single intravenous doses of 20 to 250 mcg/kg in healthy subjects and 60 mcg/kg in cancer patients, Kepivance concentrations declined over 95% in the first 30 minutes post-dose. A slight increase or plateau in concentration occurred at approximately 1 to 4 hours, followed by a terminal decline phase. Kepivance exhibited linear pharmacokinetics with extravascular distribution. In cancer patients compared with healthy subjects, after a 60 mcg/kg single dose of Kepivance the average total body clearance (CL) was 2- to 4-fold higher, and volume of distribution at steady state (Vss) was 2-fold higher. The elimination half-life was similar between healthy subjects and cancer patients (average 4.5 hours with a range of 3.3 to 5.7 hours). No accumulation of Kepivance occurred after 3 consecutive daily doses of 20 and 40 mcg/kg in healthy subjects or 60 mcg/kg in cancer patients. Age (1 to 16 years) did not affect the pharmacokinetics of palifermin over the dose range of 40 to 80 mcg/kg [see Use in Specific Populations ( 8.4)] .
Drug Interactions
Co-administration with Heparin
The potential pharmacokinetic interaction between palifermin and heparin was evaluated in a single-dose study in 27 healthy subjects receiving palifermin (60 mcg/kg) co-administered with and without therapeutic levels of unfractionated heparin. This co-administration resulted in a 5-fold increase in palifermin AUC and an 80% decrease in the mean CL. There was no significant effect of palifermin on heparin activity with respect to activated partial thromboplastin time (aPTT). A second study was conducted in 31 evaluable healthy subjects receiving palifermin (40 mcg/kg/day for 3 days) co-administered with and without therapeutic levels of unfractionated heparin. In this study, coadministration of heparin and palifermin resulted in a 425% increase in palifermin AUC and a 76.5, 73.1, and 38.8% decrease in palifermin CL, volume of distribution, and half-life, respectively. These changes in palifermin PK did not have a noticeable effect on Ki67 expression in buccal biopsies, used as a marker of epithelial cell proliferation.
Pharmacokinetics in Specific Populations
Renal Impairment
Results from a pharmacokinetics study in 24 subjects with varying degrees of renal impairment demonstrated that renal impairment has little or no influence on Kepivance pharmacokinetics.
Hepatic Impairment
The pharmacokinetic profile of patients with hepatic insufficiency has not been assessed.
Elderly
In a single-dose study, subjects received a 180-mcg/kg or 90-mcg/kg dose of palifermin administered by intravenous bolus injection. Subjects over the age of 65 (n=8) had an approximately 30% lower rate of CL on average than those 65 and younger (n=19). No dose adjustment is recommended for the geriatric population [see Use in Specific Populations ( 8.5)] .
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Palifermin was not carcinogenic in a 26-week study in rasH2 transgenic mice at intravenous doses of 0.1, 1, or 10 mg/kg/dose.
Palifermin was not genotoxic in the reverse mutation bacterial (Ames) test, cytogenic test, or rat micronucleus assay.
In a fertility and early embryonic development study, palifermin was administered intravenously to male and female rats at doses of 100, 300 or 1000 μg/kg/day. Males were dosed 28 days prior to mating through cohabitation and females were dosed 14 days prior to mating through gestation Day 7. Decreased epididymal sperm counts, and increased post-implantation losses were observed at doses ≥ 300 μg/kg/day (5 fold the recommended human dose, on a body weight basis). Increased pre-implantation loss and decreased fertility index, were observed at a palifermin dose of 1000 μg/kg/day.
14. Clinical Studies
14.1 Autologous transplantation preparative regimens that include total body irradiation
The safety and efficacy of Kepivance in decreasing the incidence and duration of severe oral mucositis in patients with hematologic malignancies (NHL, Hodgkin's disease, acute myeloid leukemia, acute lymphoblastic leukemia, chronic myeloid leukemia, chronic lymphocytic leukemia, or multiple myeloma) receiving myelotoxic therapy requiring hematopoietic stem cell support, were established in a randomized placebo-controlled clinical trial of 212 patients (Study 1) and a randomized, schedule-ranging, placebo-controlled clinical trial of 169 patients (Study 2).
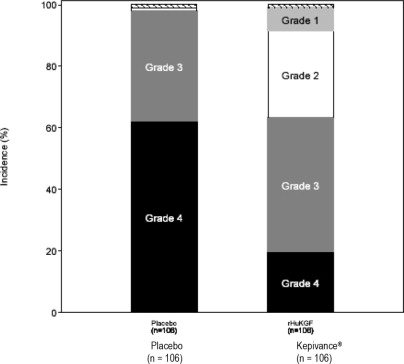
In Study 1, patients received high-dose cytotoxic therapy consisting of fractionated total-body irradiation (TBI) (12 Gy total dose), high-dose etoposide (60 mg/kg), and high-dose cyclophosphamide (100 mg/kg) followed by hematopoietic stem cell support. Patients were randomized to receive either Kepivance (n = 106) or placebo (n = 106). Kepivance 60 mcg/kg was administered as a daily intravenous injection for 3 consecutive days prior to initiation of cytotoxic therapy and for 3 consecutive days following infusion of hematopoietic stem cells. The major efficacy outcome was the number of days during which patients experienced severe oral mucositis (Grade 3/4 on the WHO [World Health Organization] scale) 1. Other analyses included the incidence, duration, and severity of oral mucositis and the use of opioid analgesia. There was no evidence of a delay in time to hematopoietic recovery in patients who received Kepivance as compared to patients who received placebo. The results of Study 1 are presented in Table 2and Figure 1.
|
* P < 0.001 compared to placebo, using Generalized Cochran-Mantel-Haenszel (CMH) test stratified for study center.
|
||
| Efficacy Variable | Kepivance
(60 mcg/kg/day) (n = 106) |
Placebo (n = 106) |
| Median (25
th, 75
thpercentile) Days of WHO Grade 3/4 Oral Mucositis*
| 3 (0, 6)
| 9 (6, 13)
|
| Incidence of WHO Grade 3/4 Oral Mucositis
| 63% (67/106)
| 98% (104/106)
|
| Median (25
th, 75
thpercentile) Days of WHO Grade 3/4 Oral Mucositis in Affected Patients
| 6 (3, 8)
(n = 67) | 9 (6, 13)
(n = 104) |
| Incidence of WHO Grade 4 Oral Mucositis
| 20%
| 62%
|
| Median (25
th, 75
thpercentile) Cumulative Opiod Dose (morphine mg equivalents)
| 212 (3, 558)
| 535 (269, 1429)
|
Figure 1: Study 1 Incidence of Oral Mucositis by Maximum Grade WHO Oral Mucositis Scale
Study 2 was a randomized, multi-center, placebo-controlledtrial comparing varying schedules of Kepivance. All patients received high-dose cytotoxic therapy consisting of fractionated TBI (12cGy total dose), high-dose etoposide (60 mg/kg), and high-dose cyclophosphamide (75-100 mg/kg) followed by hematopoietic stem cell support. The results for Study 1 were supported by results observed in the subset of patients in Study 2 who received the same dose and schedule of Kepivance administered in Study 1. One arm of Study 2 that included patients who received Kepivance for 3 consecutive days prior to initiation of cytotoxic therapy, a dose given on the last day of TBI prior to etoposide, and for 3 consecutive days following infusion of hematopoietic stem cells was prematurely closed by the Safety Committee for lack of efficacy and a trend towards increased severity and duration of oral mucositis as compared to placebo-treated patients. The Safety Committee attributed the safety finding to Kepivance having been administered within 24 hours of chemotherapy, which resulted in an increased sensitivity of the rapidly dividing epithelial cells in the immediate post-chemotherapy period [seeDosage and Administration( 2.1) and Drug Interactions ( 7)] .
- 1
- WHO Oral Mucositis Scale: Grade 1 = soreness/erythema; Grade 2 = erythema, ulcers, can eat solids; Grade 3 = ulcers, requires liquid diet only; Grade 4 = alimentation not possible.
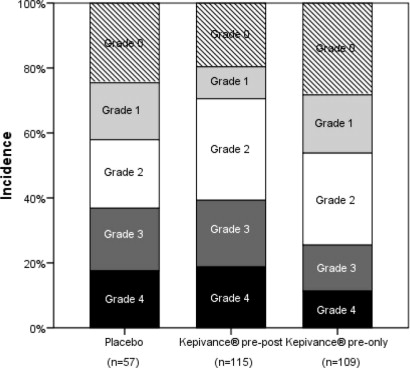
14.2 Lack of Efficacy: Autologous transplantation preparative regimen using high dose melphalan
In a post approval study, Study 3, designed to determine the efficacy of Kepivance with a high dose melphalan preparative regimen, patients with multiple myeloma were evaluated in a multicenter, randomized, double-blind, placebo-controlled trial. The conditioning regimen was melphalan (200 mg/m 2) on day -2 followed by autologous hematopoietic stem cell support. A total of 281 patients were randomized to 3 arms: Kepivance before melphalan on days -6, -5, -4 and after melphalan on days 0, 1, and 2 (pre-post) (n=115); Kepivance before melphalan on days -6, -5, -4 (pre) (n=109); or placebo (n=57).
The main outcome of the study was maximum severity of WHO oral mucositis. The median age of enrolled patients was 57 years (range 32-69), and 55% were male. The results are presented in
Figure 2. The prespecified primary analysis was a comparison between the Kepivance pre-post and pre arms to placebo. The incidence of WHO Grade 3 and 4 in the Kepivance pre-post arm was 38%, compared to 37% in the placebo arm. There were no significant differences between either of the Kepivance regimens and the placebo arm in the incidence of severe oral mucositis.
Figure 2: Incidence of Oral Mucositis by Maximum Grade WHO Oral Mucositis Scale in High Dose Melphalan Study
A subset of subjects enrolled in the multiple myeloma study were included in an evaluation for the risk of cataract development in patients receiving Kepivance treatment. Ophthalmologic examinations were performed on 101 patients enrolled in a double-blind, randomized, placebo-controlled study of two different schedules of Kepivance (pre and post chemotherapy and pre chemotherapy only) for reduction in severity of oral mucositis in subjects with multiple myeloma receiving high dose melphalan followed by autologous peripheral blood stem cell transplantation. For the primary cataract endpoint of incidence of cataract development or cataract progression at Month 12, there was a greater proportion of subjects that experienced cataract development in the Kepivance group: 48% (25/52) compared with the placebo group: 29% (4/14) (difference of 17 [95% CI: -11, 46]) [see Adverse Reactions ( 6.1)] .
14.3 Lack of Efficacy: Allogeneic Transplantation
In a post approval study, designed to determine the efficacy of Kepivance in decreasing the incidence of severe acute graft versus host disease (aGVHD) in patients with hematologic malignancies undergoing allogeneic transplantation, the incidence, duration and severity of oral mucositis was also measured. Multiple conditioning regimens were used. Patients were randomized to placebo (n=78) or to Kepivance (n=77) 60 micrograms/kg for 3 doses prior to the conditioning regimen and 180 micrograms/kg at least 24 hours from the last dose of chemotherapy and at least 24 hours before the first dose of post transplant methotrexate. There was no difference in the incidence of severe aGVHD Kepivance (16%) compared to placebo (17%). In addition to the lack of efficacy in preventing severe aGVHD, the incidence of WHO grade 3 and 4 mucositis was nominally higher in patients treated with Kepivance (81%) compared to placebo (73%).
16. How is Kepivance supplied
Kepivance is supplied as a lyophilized powder in single-dose vials containing 5.16 mg of palifermin.
Kepivance vials are supplied in:
- a dispensing pack containing 3 vials (NDC 66658-113-03)
- a dispensing pack containing 6 vials (NDC 66658-113-06)
Store Kepivance vials in the dispensing pack in its carton refrigerated at 2° to 8°C (36° to 46°F) until time of use. Protect from light.
17. Patient Counseling Information
Advise patients to report the following to healthcare providers:
- Rashes and reddening of skin [see Adverse Reactions ( 6.1)]
- Itchiness [see Adverse Reactions ( 6.1)]
- Swelling of tongue [see Adverse Reactions ( 6.1)]
- Changes in mouth and tongue sensation [see Adverse Reactions ( 6.1)]
- Alteration in taste [see Adverse Reactions ( 6.1)]
Inform patients
- That the safety and efficacy of Kepivance have not been established in patients with non-hematologic malignancies [see Indications and Usage ( 1) and Warnings and Precautions ( 5.1)]
- Of the evidence of tumor growth and stimulation in cell culture and in animal models of non-hematopoietic human tumors [see Warnings and Precautions ( 5.1) and Clinical Pharmacology ( 12.1)]
sobi
SWEDISH ORPHAN BIOVITRUM
Manufactured by
:
Swedish Orphan Biovitrum AB (publ)
SE-112 76 Stockholm, Sweden
U.S. License No. 1859
© Swedish Orphan Biovitrum AB (publ). All rights reserved.
Principal Display Panel – 3 Pack 5.16 mg/1.2 mL Carton Label
sobi
3 x 5.16 mg/vial Single Use Vials
NDC 66658-113-03
Kepivance®
(palifermin)
For Injection
5.16 mg/vial
For Intravenous Injection Only
Each Dispensing Pack contains 3 single-use vials. Each single-use
vial of Kepivance® contains 5.16 mg palifermin, 41 mg mannitol,
21 mg sucrose, 1.60 mg L-histidine, and 0.11 mg polysorbate 20
with a pH of 6.5 when reconstituted.
After reconstitution with 1.2 mL of Sterile Water for Injection, USP, the
final concentration is 5 mg/mL.
No preservatives.
Discard any unused portion.
Rx Only

Principal Display Panel – 6 Pack 5.16 mg/1.2 mL Carton Label
sobi
6 x 5.16 mg/vial Single Use Vials
NDC 66658-113-06
Kepivance®
(palifermin)
For Injection
5.16 mg/vial
For Intravenous Injection Only
Each Dispensing Pack contains 6 single-use vials. Each
single-use vial of Kepivance® contains 5.16 mg palifermin,
41 mg mannitol, 21 mg sucrose, 1.60 mg L-histidine, and
0.11 mg polysorbate 20 with a pH of 6.5 when reconstituted.
After reconstitution with 1.2 mL of Sterile Water for Injection,
USP, the final concentration is 5 mg/mL.
No preservatives.
Discard any unused portion.
Rx Only

| KEPIVANCE
palifermin injection, powder, lyophilized, for solution |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| Labeler - Swedish Orphan Biovitrum AB (publ) (354010589) |
Biological Products Related to Kepivance
Find detailed information on biosimilars for this medication.
More about Kepivance (palifermin)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Side effects
- Dosage information
- During pregnancy
- FDA approval history
- Drug class: miscellaneous uncategorized agents
- Breastfeeding
- En español